

Lecanemab: A New Alzheimer’s Drug Brings Hope and Sparks Controversy
Charlotte Tobin
Illustrations by Querida Alleyne

Most of us take for granted that we will always recognize the people we love, and few things are as difficult as being forgotten by someone who knew you well. Unfortunately, for millions of Americans, Alzheimer’s disease makes this a painful reality of everyday life. Alzheimer’s is a progressive brain disorder that not only erases memory but erodes cognition and independence more broadly — and it is extremely common. One in 10 Americans over the age of 65 lives with the disease, and as the U.S. population ages, this number is expected to grow [1]. Despite its severity and prevalence, the disease remains poorly understood [2]. Because linking its observed biological features to cognitive decline is difficult, the root cause of Alzheimer’s has several competing explanations [2]. Consequently, while existing medications treat symptoms such as irritability, inattention, and language impairment, no medication had been widely approved to address any single potential root cause of Alzheimer’s — until lecanemab (brand name Leqembi) in 2023 [3]. Approved in more than twenty countries, lecanemab represents hope for a new generation of therapies that slow the progression of Alzheimer’s rather than just manage its symptoms [4, 5]. At the same time, lecanemab’s approval has stirred debate. Uncertainty about whether its clinical benefits justify its costs and risks has raised deeper questions about how we research, evaluate, and approve treatments for complex and devastating diseases.
Fundamentals of Alzheimer's: What We Know
Alzheimer’s disease is a progressive neurodegenerative disorder, meaning it is characterized by the gradual death of neurons [6]. Neurons are specialized brain cells responsible for processing information and transmitting signals to other neurons [7]. More specifically, these cells send and receive information across small gaps between them called synapses [8]. It is the organization of these neurons and synapses into larger networks that enables core processes such as memory, learning, and behavior [9]. In the case of Alzheimer’s, synapses deteriorate and neurons die over time, progressively disrupting entire neural networks and driving the cognitive decline typically seen in individuals with the disease [10]. In early stages, symptoms manifest as minor behavioral changes, including mild confusion, difficulty concentrating, and wandering [4]. In later stages, cognitive impairment compromises the body’s most involuntary functions such as speech, movement, and swallowing [10]. Ultimately, people with Alzheimer’s can no longer control basic bodily functions and lose the ability to manage daily life on their own [4].
What Lies Behind Neurodegeneration? The Common Hypotheses
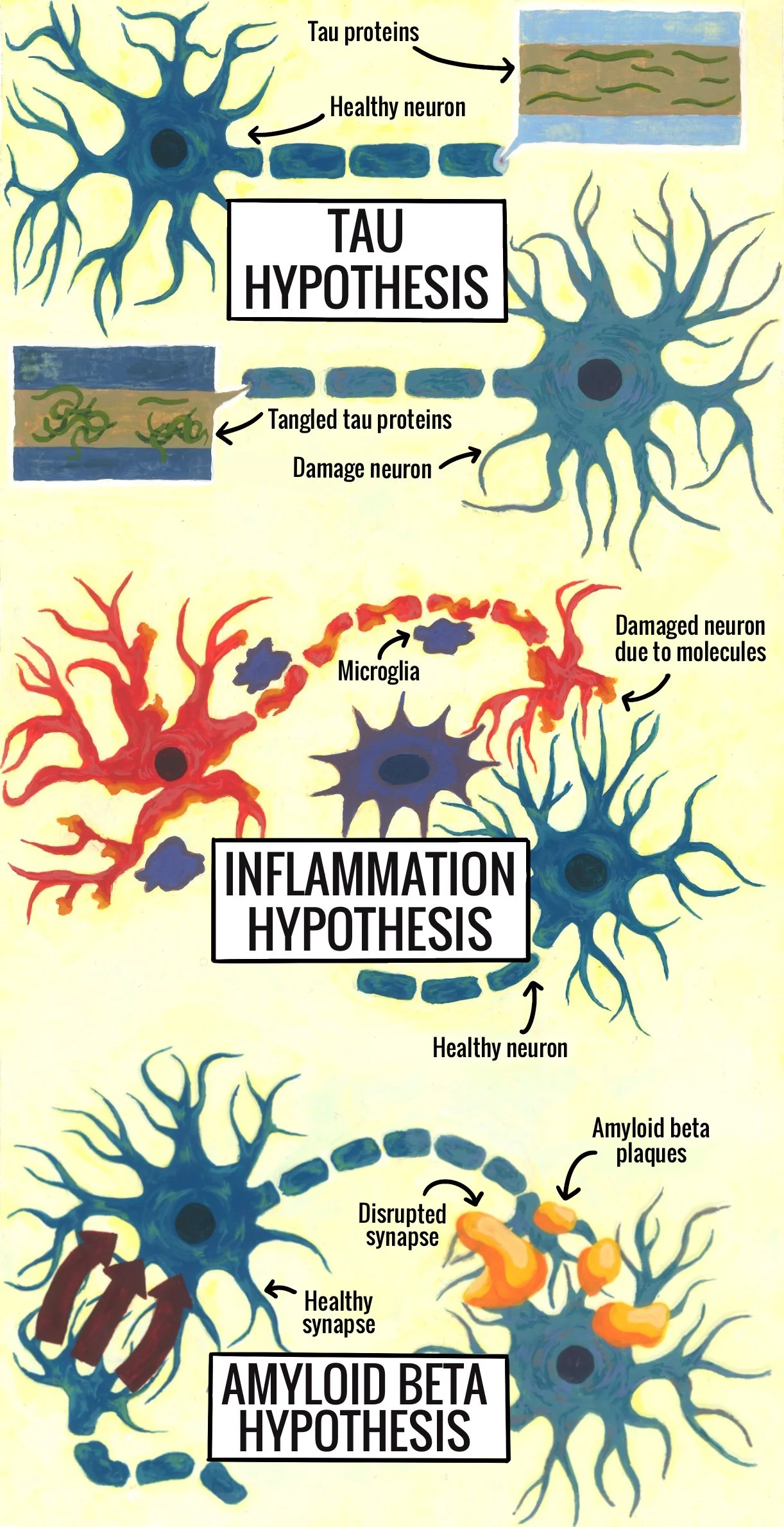
Despite the prevalence and severity of Alzheimer’s, research on the many hallmark features observed in the brains of affected individuals has yet to reveal a ‘smoking gun’ — the single biological mechanism responsible for the neuronal damage that characterizes the disease [11]. As a result, several competing hypotheses identify various proteins as the culprits of cognitive decline. One prominent explanation is the tau hypothesis, which links the irregular buildup of tau proteins — structural proteins found in neurons — to disruptions in neuronal function that can result in cell death [12]. In healthy neurons, tau stabilizes structures that support nutrient and signal transport within the cell [13]. In Alzheimer’s, tau forms tangled clumps that interfere with this transport system, damaging neurons and impairing brain function [14]. The tau hypothesis posits that these tangles are the main cause of cognitive decline [14]. Despite being a strong theory, no developed tau-targeting medication has demonstrated robust or consistent enough cognitive benefit in clinical trials to be approved [15, 16].
Another leading theory, the inflammation hypothesis, attributes neuronal damage and disease progression to the overactivation of microglia, a type of immune cell [15]. Under normal conditions, the primary function of microglia is to protect neurons by pruning inactive synapses and removing harmful debris [17]. The inflammation hypothesis maintains that the primary source of cognitive decline in Alzheimer’s is not simply the buildup of proteins like tau, but the subsequent and potentially destructive immune response these proteins elicit [18]. Within this framework, when microglia are overactive, they release inflammatory molecules that can damage surrounding neurons and exacerbate disease progression [15]. However, there is disagreement over whether this inflammation is a cause of Alzheimer’s or rather a reaction to existing damage [18]. This uncertainty, coupled with inconsistent results in clinical trials, has so far prevented any microglia-targeting therapy from reaching approval [11].
Finally, and most relevant to lecanemab, is the amyloid beta (Aß) hypothesis. During normal neuronal maintenance, the breakdown of large proteins produces Aß, a type of protein fragment [19]. While a moderate amount of Aß is healthy, an excess can accumulate into harmful clumps called plaques [20]. When these plaques form between neurons, they can contribute to the structural damage of synapses and interfere with neuron-to-neuron signaling [21]. As this damage worsens, synapses become inactive and are removed by the immune system [22]. This decrease in synapses and the resulting compromised communication are strongly linked to the cognition and memory deficits present in individuals with Alzheimer’s disease [23]. Since Aß plaques are considered a leading cause of Alzheimer’s, they are a common focus for medication aimed at addressing the root cause of the disease [24]. Among currently prescribed therapies, lecanemab was the first FDA-approved treatment shown to target this underlying mechanism of Alzheimer’s [25, 26].
How Lecanemab is Breaking the Mold
After decades of symptom-focused drug development, lecanemab belongs to a new generation of therapies designed to target a potential underlying cause of Alzheimer’s. More specifically, lecanemab primarily targets protofibrils –– smaller, earlier clumps of Aß that form as precursors to larger plaques [3]. Crucially, protofibrils are considered more toxic to neurons than plaques, and impair synaptic function early in the disease process [27]. Think of this process as mold growing in a house: By the time mold accumulates into large, visible patches, the fungus has already been silently causing irreversible structural damage to the home for much longer. Those large patches are similar to Aß plaques weakening neural networks in the brain [28]. The earlier, more inconspicuous damage is similar to the damage caused by protofibrils, which interfere with synaptic connections between neurons before any visible accumulation occurs [2,29]. Lecanemab is designed to target these early clusters and clear the larger plaques, intervening at both stages of Aß buildup and theoretically improving its effectiveness at slowing disease progression [3, 29].
This intervention works through lecanemab’s design as an antibody with the ability to target harmful amyloid structures in patients diagnosed with early-stage Alzheimer’s [30]. Antibodies are proteins that recognize and bind to specific targets in the body, effectively flagging them for removal [31]. In this case, lecanemab antibodies bind to Aß species of varying sizes and have a high affinity for the smaller toxic protofibrils [29]. By attaching to these structures, lecanemab recruits immune cells to clear the flagged amyloid from neural tissue [32]. Targeting Aß protein buildup in its earlier, more toxic form may help limit initial synapse damage, giving lecanemab the potential to slow cognitive decline at its source [33]. Because Aß accumulation is one of the most characteristic features of Alzheimer’s, targeting the clumps has long been an area of interest for researchers [33]. To translate lecanemab from a theory to tangible benefit for patients, the drug was evaluated in a series of clinical trials [29]. The results told an interesting story.
Evaluating the Evidence
Lecanemab was tested in a clinical study of several hundred people with early-stage Alzheimer’s during which each participant received either lecanemab or a placebo for a year and a half [29]. In order to be considered successful, lecanemab could not just perform better than the placebo; it had to work significantly better as measured by a sensitive composite cognitive scale [34]. At one year, though the treatment group experienced less cognitive decline than the placebo, the trial failed to meet the exact requirements for the sensitive measurement [34, 35]. The study continued for an additional six months, at which point the researchers performed a traditionally used though less precise analysis and found that lecanemab reduced cognitive decline by 27% compared to the placebo [26]. On top of this reduction, the treatment group showed less Aß buildup by the end of the study, meaning the drug worked as intended in the brain [26]. Because lecanemab slowed cognitive decline and reduced amyloid levels with relatively few adverse reactions, it was uniquely successful compared to past clinical trials of similar therapies at the time [26, 36]. The benefits of lecanemab led to its full Food and Drug Administration (FDA) approval in July 2023 [36].
To understand this approval, it is important to examine how the FDA evaluates new therapies. In the United States, all drug approval decisions are made by the FDA, often with input from independent advisory committees consisting of various experts in relevant fields alongside patient and consumer representatives [37]. The FDA follows the advisory committee’s recommendation the vast majority of the time [38]. One notable exception was aducanumab (Aduhelm), an earlier Aß-targeting therapy that was approved despite near-unanimous advisory committee opposition; it was discontinued shortly after amid widespread controversy, becoming one of the most contentious decisions in recent FDA history [38]. Evaluating Alzheimer’s medications is notoriously difficult, which is part of what made lecanemab’s approval so notable and prior decisions so fraught. Because the underlying biological processes behind Alzheimer’s remain uncertain, it is hard to reach an agreement on whether any given biological improvement, like amyloid reduction, is sufficient to produce meaningful clinical benefit [26]. In situations of unmet need, where a disease is severe and treatment options are limited, regulators are willing to tolerate more uncertainty about a drug’s long-term benefits even if a therapy’s success is merely plausible and not proven [39]. These approvals reflect the challenge of defining a tolerable level of uncertainty and set a precedent for future medications. Researchers may disproportionately pursue Aβ-targeting treatments because doing so seems more likely to succeed given past approvals. As a result, there is concern that alternative approaches that could prove even more valuable will be ignored [40].
Lecanemab or Lecanemaybe? The Enduring Controversy
An additional complication to the scientific debate surrounding the Aβ hypothesis is that the levels of Aβ observed in the brain do not consistently align with the severity of cognitive symptoms [41]. While the accumulation of Aβ is considered to be a major contributor to cognitive decline, some individuals exhibit levels of buildup typically associated with late-stage impairment while remaining at their cognitive baseline. This apparent ‘resistance’ to neurodegeneration suggests that plaques themselves may not cause the decline in symptoms but may rather be a tightly linked result of the true underlying mechanism of Alzheimer’s [41]. This complexity further calls into question whether removing Aβ alone is sufficient to meaningfully alter disease progression and also helps explain why proven reductions in Aβ accumulation do not consistently translate to substantial clinical improvement.
A specific complicating factor in evaluating lecanemab’s effectiveness is the widely cited ‘27%’ reduction in cognitive decline, which was obtained using a less precise though more conventional overall cognitive scale [26, 42]. At first glance, a 27% reduction seems like a substantial effect. However, this figure is misleading because it reflects a relative difference between the treatment and placebo group, rather than the actual magnitude of change experienced by patients [26]. The difference between the amount the treatment group declined relative to theother group was 27%, but relative to the overall cognitive scale was just 2.5%. It’s like saying losing $7 in the second round of poker is a 30% improvement compared to losing $10 in the first round — technically true but not necessarily meaningful in practice. Furthermore, in order for there to continue to be a 27% difference, the gap in cognitive decline between the placebo and treatment groups must continue to grow over time. If both groups decline at similar rates after the trial period, the relative benefit of lecanemab compared to placebo will not hold, suggesting the reported effect may be an overestimate of long-term impact [36, 42].
Weighing the Risks: The Side Effects and Burdens of Treatment
The uncertainties surrounding lecanemab’s effectiveness become even more important when considering the realities of the treatment, as it is an intervention that requires continual monitoring, carries nontrivial risk, and may be massively burdensome for both caregivers and increasingly incapacitated individuals [36]. Lecanemab is administered twice-monthly and comes with a black box warning, the FDA’s strongest safety designation, signaling a risk of potentially life-threatening side effects [43]. One of the most notable side effects is an increased risk of amyloid-related imaging abnormality (ARIA), a finding on brain imaging that is associated with swelling or bleeding [26, 36]. Though ARIAs are often mild or asymptomatic, patients may experience headaches, confusion, vomiting, vision loss, difficulty walking, and even death [44]. The association of lecanemab with the potentially severe risks of ARIAs is the reason for the drug’s black box warning [45]. While ARIAs occurred in both treatment and placebo groups, they occurred at higher rates in the treatment group and represent a distinct risk that patients should weigh when considering lecanemab [26].
ARIA prevalence is not an isolated burden; it also necessitates frequent imaging, adding to the overall hardship of treatment [36]. Magnetic resonance imaging (MRI) frequencies vary from patient to patient depending on risk factors, notably increased genetic risk for serious adverse events [46]. Regardless of results from genetic pre-screening, at least four MRIs are required in the first six months of lecanemab injections. Patients may need additional MRIs if they develop ARIA, which was the case in a significant portion of the clinical trial patients [36]. The cost of this imaging is not included in the drug’s $26,500 annual cost [47]. For patients and caregivers already navigating a difficult diagnosis, this adds both financial strain and the burden of frequent medical appointments [48]. And, without infrastructure to reduce these practical burdens, sustained treatment will remain out of reach for many, limiting how much lecanemab can meaningfully improve lives beyond a clinical setting [49].
Embracing Complexity and Looking Forward
The evidence surrounding lecanemab is difficult to evaluate. On the one hand, treatment reduces the amount of Aβ plaque buildup [26, 50]. On the other hand, the translation of that neurophysical reduction to a distinct cognitive benefit is modest [49]. Furthermore, the drug’s mechanism rests on the Aβ hypothesis, which remains scientifically contested [51]. Though complicated, the successes and shortcomings of lecanemab contribute to our understanding of how the FDA recalibrates evidence thresholds in the face of unmet need — a topic undoubtedly important to understand, with many implications for future therapies and other diseases [52]. The case also spurs questions about acceptable levels of treatment burden, a question that is perhaps not as commonly asked but is nonetheless an important consideration [52].
Importantly, the story of lecanemab speaks to the limitations of relying on a single mechanism to explain and treat a disease as complex as Alzheimer’s [36]. The body and brain are a complex system, reflecting constant interactions between many biological mechanisms. Prioritizing one explanation for a condition over another risks narrowing our understanding and curtailing meaningful progress [51]. Lecanemab should be seen as a component of a broader therapeutic strategy that addresses more than just a single mechanism. Single-target approaches are inherently limited, and lecanemab should not be considered the ‘final boss’ of Alzheimer's therapies [36]. Instead, a combination of therapies must be investigated alongside broader considerations of lifestyle and environment to fully and effectively address the onset and progression of the disease [36, 53]. Ultimately, Alzheimer’s may not demand a single answer, but rather a willingness to accept complexity both in how it is studied and how it is treated.
References
ALZHEIMER'S ASSOCIATION REPORT. (2025). 2025 Alzheimer's disease facts and figures. Alzheimer’s & Dementia, 21(4). doi:10.1002/alz.70235.
Hampel, J., Hardy, J., Belnnow, K., Chen, C., Perry, G., Hyun Kim, S., Villemagne, V. L., Aisen, P., Vendruscolo, M., Iwatsubo, T., Masters, C. L., Cho, M., Lannfelt, L., Cummings, J. L., & Vergallo, A. (2021). The amyloid-β pathway in Alzheimer’s disease. Molecular Psychiatry, 26, 5481–5503. doi:10.1038/s41380-021-01249-0.
Cummings, J., Leisgang O., Amanda M., Cammann, D., Powell, J., & Chen, J. (2024). Anti-amyloid monoclonal antibodies for the treatment of Alzheimer's disease. BioDrugs, 38, 5-22. doi: 10.1007/s40259-023-00633-2.
Alzheimer's Association Report. (2024). 2024 Alzheimer's disease facts and figures. Alzheimer's and Dementia, 20(5), 3708-3821. doi: 10.1002/alz.13809.
Yakushev, I., Verger, A., Brendel, M., Cecchin, D., Fernandez, P. A., Fraioli, F., Grimmer, T., Tolboom, N., Traub-Weidinger, T., Guedji, E., & Van Weehaeghe, D. (2025). Lecanemab approval in the EU: What should we be ready for? The EANM perspective. Eur J Nucl Mol Imaging, 52(5), 1607-1610. doi: 10.1007/s00259-025-07066-9.
Breijyeh, Z., & Karaman, R. (2020). Comprehensive review on Alzheimer’s disease: Causes and treatments. Molecules, 25(24), 5789. doi: 10.3390/molecules25245789.
Deco, G., & Kringelbach M. L. (2017). Hierarchy of information processing in the brain. Neuron, 94(5), 961-968. doi: 10.1016/j.neuron.2017.03028
Von Bartheld, C. S. (2018). Myths and truths about the cellular composition of the human brain: A review of influential concepts. Journal of Chemical Neuroanatomy, 93, 2-15. doi:10.1016/j.jchemneu.2017.08.004.
Dupret, D., Fusi, S., & Panzeri, S. (2026). Neural population activity for memory: Properties, computations, and codes. Neuron, 114(3), 390-407. doi:10.1016/j.neuron.2025.11.007.
Kamatham, P. T., Shukla, R., Khatri, D. K., & Vora, L. K. (2024). Pathogenesis, diagnostics, and therapeutics for Alzheimer's disease: Breaking the memory barrier. Ageing Research Reviews, 101. doi:10.1016/j.arr.2024.102481.
Zhang, J., Zhang, Y., Wang, J., Xia, Y., Zhang, J., & Chen, L. (2024). Recent advances in Alzheimer’s disease: mechanisms, clinical trials and new drug development strategies. Signal Transduction and Targeted Therapy, 9. doi:10.1038/s41392-024-01911-3.
Kametani, F., & Hasegawa, M. (2018). Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer's disease. Frontiers in Neuroscience, 12. doi:10.3389/fnins.2018.00025.
Lippens, G., & Gigant, B. (2019). Elucidating tau function and dysfunction in the era of cryo-EM. Journal of Biological Chemistry, 294(24), 9316-9325. doi:10.1074/jbc.REV119.008031.
Cáceres, C., Heusser, B., Garnham, A., & Moczko, E. (2023). The major hypotheses of Alzheimer’s disease: related nanotechnology-based approaches for its diagnosis and treatment. Cells, 12(23), 2669. doi:10.3390/cells12232669.
Du, X., Wang, X., & Geng, M. (2018). Alzheimer’s disease hypothesis and related therapies. Translational Neurodegeneration, 7(2). doi:10.1186/s40035-018-0107-y.
Bakota, L., & Brandt, R. (2026). Towards mechanism-based tau-targeted therapies. Neural Regeneration Research, 21(2), 687-688. doi:10.4103/NRR.NRR-D-24-01240.
Kierdorf, K., & Prinz, M. (2017). Microglia in steady state. Journal of Clinical Investigation, 127(9), 3201-3209. doi:10.1172/JCI90602.
Kinney, J.W., Bemiller, S.M., Murtishaw, A.S., Leisgang, A.M., Salazar, A.M. and Lamb, B.T. (2018). Inflammation as a central mechanism in Alzheimer's disease. Alzheimer's & Dementia: Translational Research & Clinical Interventions, 4, 575-590. doi:10.1016/j.trci.2018.06.014.
Sehar, U., Rawat, P., Reddy, A. P., Kopel, J., & Reddy, P. H. (2022). Amyloid beta in aging and Alzheimer’s disease. International Journal of Molecular Sciences, 23(21), 12924. doi:10.3390/ijms232112924.
Takahashi, R.H., Nagao, T. & Gouras, G.K. (2017). Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer's disease. Pathology International, 67, 185-193. doi:10.1111/pin.12520.
Zhang, H., Jiang, X., Ma, L., Wei, W., Zehui, L., Chang, S., Wen, J., Sun, J., & Li, H. (2022). Role of Aβ in Alzheimer’s-related synaptic dysfunction. Frontiers in Cell and Developmental Biology, 10. doi:10.3389/fcell.2022.964075.
Rajendran, L., & Paolicelli, R. C. (2018). Microglia-mediated synapse loss in Alzheimer's disease. Journal of Neuroscience, 38(12), 2911-2919. doi:10.1523/JNEUROSCI.1136-17.2017.
Tönnies E., & Trushina E. (2017). Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. Journal of Alzheimer’s Disease, 57(4), 1105-1121. doi:10.3233/JAD-161088.
Yadollahikhales, G., & Rojas, J. C. (2023). Anti-amyloid immunotherapies for Alzheimer's disease: A 2023 clinical update. Neurotherapeutics, 20(4), 914–931. doi:10.1007/s13311-023-01405-0.
Papaliagkas, V. (2025). Anti-amyloid therapies for Alzheimer’s disease: progress, pitfalls, and the path ahead. International Journal of Molecular Sciences, 26(19), 9529. doi:10.3390/ijms26199529.
van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., Kanekiyo, M., Li, D., Reyderman, L., Cohen, S., Froelich, L., Katayama, S., Sabbagh, M., Vellas, B., Watson, D., Dhadda, S., Irizarry, M., Kramer, L. D., & Iwatsubo, T. (2022). Lecanemab in Early Alzheimer’s disease. The New England Journal of Medicine, 388, 9-21. doi:10.1056/NEJMoa2212948.
He, Y., Wei, M., Wu, Y., Qin, H., Li, W., Ma, X., Cheng, J., Ren, J., Shen, Y., Chen, Z., Sun, B., Huang, F.-D., Shen, Y., & Zhou, Y.-D. (2019). Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nature Communications, 10, 1193. doi:10.1038/s41467-019-09114-z.
Marin, M. A., Ziburkus, J., Jankowsky, J., & Rasband, M. N. (2016). Amyloid-β plaques disrupt axon initial segments. Experimental Neurology, 281, 93–98. doi:10.1016/j.expneurol.2016.04.018.
Vitek, G. E., Decourt, B., & Sabbagh, M. N. (2023). Lecanemab (BAN2401): an anti–beta-amyloid monoclonal antibody for the treatment of Alzheimer disease. Expert Opinion on Investigational Drugs, 32(2), 89–94. doi:10.1080/13543784.2023.2178414.
Chowdhury S., & Chowdhury N.S. (2023). Novel Anti-amyloid-beta (Aβ) Monoclonal antibody lecanemab for Alzheimer’s disease: A systematic review. International Journal of Immunopathology and Pharmacology, 37. doi:10.1177/03946320231209839.
Aboul-Ella, H., Gohar, A., Ahmed Ali, A., Ismail, L.M., El-Regal Mahmoud, A.E., Elkhatib, W.F., & Aboul-Ella, H. (2024). Monoclonal antibodies: From magic bullet to precision weapon. Molecular Biomedicine, 5(47). doi:10.1186/s43556-024-00210-1.
Barrera-Ocampo, A., & Lopera, F. (2016). Amyloid-beta immunotherapy: The hope for Alzheimer’s disease? Colombia Médica, 47(4), 203-212.
Ono, K., & Tsuji, M. (2020). Protofibrils of amyloid-β are important targets of a disease-modifying approach for Alzheimer’s disease. International Journal of Molecular Sciences, 21(3), 952. doi:10.3390/ijms21030952.
Berry, D.A., Dhadda, S., Kanekiyo, M., Li, D., Swanson, C.J., Irizarry, M., Kramer, L.D., & Berry, S.M. (2023). Lecanemab for patients with early Alzheimer’s disease: Bayesian analysis of a phase 2b dose-finding randomized clinical trial. JAMA Network Open, 6(4). doi:10.1001/jamanetworkopen.2023.7230.
Swanson, C.J., Zhang, Y., Dhadda, S., Wang, J., Kaplow, J., Lai, R.Y.K., Lannfelt, L., Bradley, H., Rabe, M., Koyama, A., Reyderman, L., Berry, D.A., Berry, S., Gordon, R., Kramer, L.D., & Cummings J.L. (2021). A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, anti-Aβ protofibril antibody. Alzheimer’s Research & Therapy, 13(80). doi:10.1186/s13195-021-00813-8.
Cummings, J., Apostolova, L., Rabinovici, G.D., Atri, A., Aisen, P., Greenberg, S., Hendrix, S., Selkoe, D., Weiner, M., Petersen, R.C., & Salloway, S. (2023). Lecanemab: appropriate use recommendations. The Journal of Prevention of Alzheimer’s Disease, 10(3), 362-377. doi:10.14283/jpad.2023.30.
Corbin, J., & Walker, A.J. (2023). Chapter 75 - FDA overview. Translational Radiation Oncology, 453-457. doi:10.1016/B978-0-323-88423-5.00019-4.
Daval, J.R., Teng, T.W., & Russo, M. (2023). Association of advisory committee votes with US Food and Drug Administration decision making on prescription drugs, 2010-2021. JAMA Health Forum, 4(7). doi:10.1001/jamahealthforum.2023.1718.
Alexander, G.C., Knopman, D.S., Emerson, S.S., Ovbiagele, B., Kryscio, R.J., Perlmutter, J.S., & Kesselheim, A.S. (2021). Revisiting FDA approval of aducanumab. The New England Journal of Medicine, 385(9), 769-771. doi:10.1056/NEJMp2110468.
van Bokhoven, P., de Wilde, A., Vermunt, L., Leferink, P.S., Heetveld, S., Cummings, J., Scheltens, P., & Vijverberg, E.G.B. (2021). The Alzheimer’s disease drug development landscape. Alzheimer’s Research and Therapy, 13(186). doi:10.1186/s13195-021-00927-z.
Gómez-Isla, T., & Frosch, M.P. (2022). Lesions without symptoms: understanding resilience to Alzheimer’s disease neuropathological changes. Nature Reviews Neurology, 18, 323-332. doi:10.1038/s41582-022-00642-9.
Espay, A.J., Kepp, K.P., & Herrup K. (2024). Lecanemab and donanemab as therapies for Alzheimer’s disease: An illustrated perspective on the data. eNeuro, 11(7). doi:10.1523/ENEURO.0319-23.2024.
Rajendran, Y., Kondampati, N., Eerike M., Mali, K., & C, L.F. (2024). A longitudinal analysis of black box warnings: Trends and implications for drug safety. Cureus, 16(4). doi:10.7759/cureus.57597.
Doran, S.J., & Sawyer, R.P. (2024). Risk factors in developing amyloid related imaging abnormalities (ARIA) and clinical implications. Frontiers in Neuroscience, 18. doi:10.3389/fnins.2024.1326784.
Xing, X., Zhang, X., Wang, K., Wang, Z., Feng, Y., Li, X., Hua, Y., Zhang, L., & Dong, X. (2025). Post-marketing safety concerns with lecanemab: A pharmacovigilance study based on the FDA adverse event reporting system database. Alzheimer’s Research and Therapy, 17(15). doi:10.1186/s13195-024-01669-4.
Villain, N., Planche, V., Lilamand, M., Cordonnier, C., Soto-Martin, M., Mollion, H., Bombois, S., & Delrieu, J. (2025). Lecanemab for early Alzheimer’s disease: Appropriate use recommendations from the French federation of memory clinics. The Journal of Prevention of Alzheimer’s Disease, 12(4). doi:10.1016/j.tjpad.2025.100094.
Jiang, X., Lv, G., Mendes, M.E., Wu, J., Yuan, J., & Lu, Z.K. (2026). Lifetime cost-effectiveness of Lecanemab for early Alzheimer’s disease. Frontiers in Public Health, 14. doi:10.3389/fpubh.2026.1692508.
Anil, A.A., Krishnan, A.L., Chandra, P., & Unnikrishnan, M.K. (2025). Lecanemab: The advent of biologicals in Alzheimer’s disease, affordability, and clinical relevance. Journal of Applied Pharmaceutical Science, 15(3), 30-38. doi:10.7324/JAPS.2025.204843.
van Veluw, S.J., & Young, M.J. (2024). Ethical considerations for the use of anti-amyloid immunotherapies in patients with early Alzheimer’s disease. Alzheimer’s and Dementia, 20(5), 3664-3665. doi:10.1002/alz.13795.
Albertini, G., Zielonka, M., Cuypers, M., Snellinx, A., Xu, C., Poovathingal, S., Wojno, M., Davie, K., van Lieshout, V., Craessaerts, K., Wolfs, S., Pasciuto, E., Jaspers, T., Horré, K., Serneels, L., Fiers, M., Dewilde, M., & De Strooper, B. (2025). The Alzheimer’s therapeutic Lecanemab attenuates Aβ pathology by inducing an amyloid-clearing program in microglia. Nature Neuroscience, 29, 100-110. doi: 10.1038/s41593-025-02125-8
Behl, C. (2024). In 2024, the amyloid-cascade-hypothesis still remains a working hypothesis, no less but certainly no more. Frontiers in Aging Neuroscience, 16. doi:10.3389/fnagi.2024.1459224.
Largent, E.A., Peterson, A., & Fernandez, H. (2021). FDA drug approval and the ethics of desperation. JAMA Internal Medicine, 181(12), 1555-1556. doi: 10.1001/jamainternmed.2021.6045
Livingston, G., Huntley, J., Sommerlad, A., Ames, D., Ballard, C., Banerjee, S., Brayne, C., Burns, A., Cohen-Mansfield, J., Cooper, C., Costafreda, S. G., Dias, A., Fox, N., Gitlin, L. N., Howard, R., Kales, H. C., Kivimaki, M., Larson, E. B., Ogunniyi, A., … & Mukadam, N. (2020). Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. The Lancet, 396(10248), 413-446. doi:10.1016/S0140-6736(20)30367-6.